


:quality(80)/p7i.vogel.de/wcms/d9/13/d913edd395d2c358d596751ab3fd0dee/0132492220v1.jpeg "(Bild: VCG)")
:quality(80)/p7i.vogel.de/wcms/eb/0a/eb0af6c80446441945002939bca1dae1/0132404032v2.jpeg "Prof. Dr. Simon Ebbinghaus und Mailin Becker (r.) haben ein neues bildgebendes Verfahren entwickelt. (Bild: RUB, ehrstuhl für Biophysikalische Chemie)")
:quality(80)/p7i.vogel.de/wcms/35/e2/35e2b141e34ba3d45d19e97bb30e8502/0132382297v2.jpeg "Der Praxistag HPLC bringt Anwender zum Thema Flüssigchromatographie zusammen (Aufnahme vom Praxistag HPLC 2025). (Bild: VCG)")
:quality(80)/p7i.vogel.de/wcms/f4/9b/f49b99a6e8b33adac33a2fd63d15530a/0132188748v2.jpeg "Cryo-LA-ICP-TOFMS Setup am Alfred-Wegener-Institut (Bild: Alfred-Wegener-Institut / Pascal Bohleber)")
:quality(80)/p7i.vogel.de/wcms/d3/83/d3839fc5d7d5590eead5fd606c6479b5/0132374775v2.jpeg "Krümel und Brotreste haben schon unsere Großeltern für Paniermehl und Semmelklöße weiterverwendet. Was früher selbstverständlich war, heißt heute „Upcycled Ingredients“ und fasst in der Lebensmittelindustrie zunehmend Fuß. So wird Lebensmittelverschwendung reduziert. (Symbolbild) (Bild: frei lizenziert, Delfina Cocciardi)")
:quality(80)/p7i.vogel.de/wcms/c9/22/c9229cc360d30a8dce68bf4a6fc6bd98/0131564364v2.jpeg "Taschen-pH-Meter Five Go F2 (Bild: Carl Roth)")
:quality(80)/p7i.vogel.de/wcms/c7/32/c7324b06bc935a3073609e58b42ba02b/0132559719v1.jpeg "Ein internationales Forschungsteam hat erstmals identifiziert, welche Bakterien in der Stallluft für den sogenannten „Bauernhof-Effekt“ verantwortlich sind, der vor Allergien, Asthma und Heuschnupfen schützen kann. (Symbolbild) (Bild: © stefano - stock.adobe.com)")
:quality(80)/p7i.vogel.de/wcms/3e/19/3e19a8240deeac0e31a919d21738aef4/0132546548v2.jpeg "Pascal Daloz, Chief Executive Officer und Vorstandsvorsitzender von Dassault
Systèmes: „Die Life-Sciences-Branche tritt in eine neue Ära der Innovation ein – angetrieben durch künstliche Intelligenz und den Wandel hin zur Präzisionsmedizin –, während sie gleichzeitig mit zunehmender operativer Komplexität und strengeren regulatorischen Anforderungen konfrontiert ist.“ (Bild: Dassault Systèmes)")
:quality(80)/p7i.vogel.de/wcms/4b/bd/4bbd462ba9c312123106b29578bf8d3e/0132497884v2.jpeg "Gemeinsam mit ihrem Forschungsteam wollen Dr. Henning Ortkraß und Janina Niemeyer das Lightweaver-Mikroskop auf den Markt bringen. (Bild: Jana Haver)")
:quality(80)/p7i.vogel.de/wcms/14/16/141629871c69e4d75dd778572abbfc4e/image1-664x374v1.jpeg "Am Rand einer Landstraße hat das Projektteam Bodenproben genommen, um sie auf Mikroplastik zu untersuchen. (Bild: Prof. Dr. Sebastian Hein / Hochschule für Forstwirtschaft Rottenburg)")
:quality(80)/p7i.vogel.de/wcms/1b/08/1b08fc824322a8f98d25c546e0b54fa9/15566-d3fr7acb-1920x1079v1.jpeg "Detailaufnahme eines geschnittenen Kieselholzes vom Kyffhäuser mit einem horizontalen Durchmesser von ca. 20 Zentimetern. Die weiß-grauen Flecken sind verschiedene helle und dunkle Quarzgenerationen. (Bild: © Steffen Trümper)")
:quality(80)/p7i.vogel.de/wcms/d3/26/d3267fde90e79df9be09c83b4401448c/5u3a4062-6720x3778v1.jpeg "Die Empa-Forschenden Fiorella Pitaro und Bernd Nowack entwickeln Methoden, mit denen sich Sicherheit und Nachhaltigkeit von neuen Chemikalien ermitteln lassen. (Bild: Empa)")
:quality(80)/p7i.vogel.de/wcms/2e/17/2e1795b6750cc3eaad6ba81c0bc79d16/0100811496v7.jpeg "Aktuelle Nachrichten aus Labortechnik, Pharmaindustrie und den Naturwissenschaften (Bild: ©viperagp - stock.adobe.com)")
Datenanalyse mit Künstlicher Intelligenz Proteomics: 100 Mal weniger Fehler – mit Maschinellem Lernen
Fortschritt für Proteomics: Mit künstlicher Intelligenz und Maschinellem Lernen ist es Forschenden der Technischen Universität München (TUM) gelungen, die massenhafte Analyse von Proteinen aus beliebigen Organismen deutlich schneller als bisher und nach eigenen Angaben praktisch fehlerfrei zu machen. Der neue Ansatz kann sowohl in der Grundlagen- als auch in der klinischen Forschung angewandt werden.
Anbieter zum Thema
:fill(fff,0)/images.vogel.de/vogelonline/companyimg/6400/6447/65.jpg "Tosoh_Bioscience_Logo.jpg ()")
:fill(fff,0)/images.vogel.de/vogelonline/companyimg/86900/86921/65.jpg "LOGO.jpg ()")
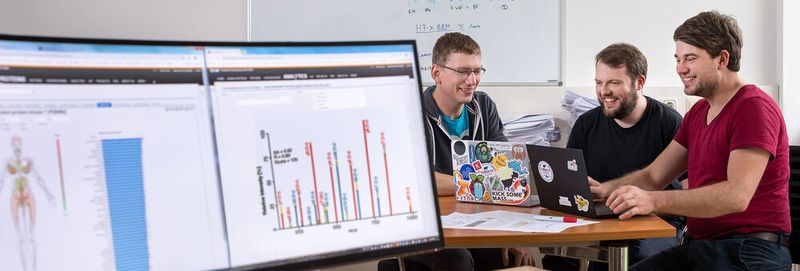
München – Das Genom jedes Organismus enthält die Baupläne für Tausende von Eiweißen, die praktisch alle Funktionen des Lebens steuern. Fehlerhafte Proteine führen zu schweren Krankheiten wie Krebs, Diabetes oder Demenz. Eiweiße sind somit auch die wichtigsten Angriffspunkte für Medikamente.
Damit man Lebensvorgänge und Erkrankungen besser verstehen und passendere Therapien entwickeln kann, muss man möglichst viele Proteine gleichzeitig analysieren. Aktuell wird hierzu die Massenspektrometrie genutzt, die in der Lage ist, Art und Menge der Eiweiße in einem biologischen System zu bestimmen. Jedoch machen die derzeitigen Verfahren der Datenanalyse noch viele Fehler.
Einem Team der TU München um den Bioinformatiker Mathias Wilhelm und den Biochemiker Bernhard Küster, Professor für Proteomik und Bioanalytik an der TU München, ist es nun gelungen, massenhaft erhobene proteomische Daten zu nutzen, um ein neuronales Netzwerk so zu trainieren, dass es Proteine deutlich schneller und praktisch fehlerfrei erkennen kann.
Proteom-Datenanalyse: Lösung für ein „gravierendes Problem“
Massenspektrometer messen Proteine nicht direkt, sondern analysieren kleinere Peptide, bestehend aus Aminosäuresequenzen mit bis zu 30 Bausteinen. Die gemessenen Spektren dieser Ketten werden mit Datenbanken abgeglichen, um sie einem bestimmten Protein zuzuordnen. Die Auswertesoftware kann jedoch nur einen Teil der enthaltenen Informationen nutzen. Daher werden manche Proteine nicht oder falsch erkannt.
„Das ist ein gravierendes Problem“, sagt Küster. Das neuronale Netzwerk, das das TUM-Team entwickelt hat, nutzt alle Informationen der Spektren für die Identifizierung. „Dadurch verpassen wir weniger Proteine und es passieren 100 mal weniger Fehler“, so Bernhard Küster.
KI-Software ist auf das Proteom aller Organismen anwendbar
„Prosit“, wie die Forscher die KI-Software nennen, ist „auf alle Organismen dieser Welt anwendbar, auch wenn man deren Proteome vorher nie untersucht hat“, sagt Mathias Wilhelm. „Das ermöglicht Untersuchungen, die vorher nicht denkbar waren.“
Der Algorithmus ist mit Hilfe von 100 Millionen Massenspektren so umfangreich angelernt worden, dass er ohne erneutes Trainieren für alle gängigen Massenspektrometer eingesetzt werden kann. „Unser System ist hier weltweit führend“, sagt Küster.
:quality(80)/images.vogel.de/vogelonline/bdb/1548600/1548687/original.jpg "Maschinelles Lernen hilft bei der Verarbeitung großer Datenmengen, braucht aber zunächst selbst viele Daten zum „Einlernen“ (Symbolbild). (gemeinfrei)")
Computertechnologie in der Wissenschaft
Wie man maschinellem Lernen auf die Sprünge helfen kann
Leistungsfähigere Geräte für einen Milliardenmarkt?
Kliniken, Biotechunternehmen, Pharmafirmen und die Forschung nutzen solche Hochleistungsgeräte; es ist schon jetzt ein Milliardenmarkt. Mit „Prosit“ können zukünftig noch leistungsfähigere Geräte entwickelt werden. Ebenso werden Forscher und Mediziner besser und schneller nach Biomarkern im Blut oder Urin von Patienten suchen oder Therapien hinsichtlich der Wirksamkeit überwachen können.
Auch für die Grundlagenforschung versprechen sich die Forscher viel. „Mit dem Verfahren kann man neuen Regulationsmechanismen in Zellen auf die Spur kommen“, so Küster. „Wir erhoffen uns hier einen erheblichen Erkenntnisgewinn, der sich mittel- und langfristig in der Behandlung von Erkrankungen von Mensch, Tier und Pflanze niederschlagen wird.“
Auch Wilhelm erwartet, dass „KI Methoden wie Prosit schon bald das Forschungsfeld der Proteomik nachhaltig verändern werden, da sie in nahezu allen Bereichen der Proteinforschung eingesetzt werden können.“
Originalpublikation: Siegfried Gessulat, Tobias Schmidt, Daniel Paul Zolg, Patroklos Samaras, Karsten Schnatbaum, Johannes Zerweck, Tobias Knaute, Julia Rechenberger, Bernard Delanghe, Andreas Huhmer, Ulf Reimer, Hans-Christian Ehrlich, Stephan Aiche, Bernhard Küster und Mathias Wilhelm: Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning; Nature Methods, 27.05.2019 – DOI: 10.1038/s41592-019-0426-7
(ID:45952090)
:quality(80)/p7i.vogel.de/wcms/43/0e/430e4c2529fb31bdb5c2500f04ed707c/0130440929v2.jpeg "TUM-Forscher Dr. Kun Sun hält eine Perowskit-Solarzelle in der Hand. (Bild: Dr. Yuxin Liang / TUM)")
:quality(80)/p7i.vogel.de/wcms/17/1c/171c35df5f9bc0c1d1a399461e46f993/0126322582v1.jpeg "Prof. Dr. Albert Sickmann (li.) und Prof. Dr. Matthias Gunzer (re.) (Bild: ISAS)")